
Hypofosfatázie a X-vázaná hypofosfatemická křivice v dětském věku
Může praktický zubní lékař pomoci v časné diagnostice?
Abstrakt: Hypofosfatázie (HPP) je vzácné onemocnění způsobené nedostatečnou produkcí alkalické fosfatázy (ALP). Dochází k demineralizaci skeletu se závažnými extraskeletálními symptomy včetně postižení dentice, a to k předčasné ztrátě dočasných zubů ve frontálním úseku čelistí. Diagnostika je založena na nízké koncentraci ALP v séru v kombinaci s typickými klinickými projevy. Od roku 2015 je dostupná substituční enzymatická léčba pomocí rekombinantní ALP.
X-vázaná hypofosfatemická křivice (XLH) je nejčastějším projevem křivice a osteomalacie. Projevuje se růstovou retardací, deformitami dolních končetin, bolestí svalů a kostí. Dentální manifestace se projevuje dysplazií dentinu s tvorbou abscesů u intaktních zubů, především v dočasné dentici. Léčba spočívá v pravidelné aplikaci protilátky proti FGF23 (fibroblastový růstový hormon 23), burosumabu. Burosumab je rekombinantní lidská monoklonální protilátka (IgG1), která se váže na FGF23 a tlumí jeho aktivitu.
Abstract: Hypophosphatasia (HPP) is a rare disease caused by insufficient production of alkaline phosphatase. Main symptoms are skeletal demineralisation and severe extra-skeletal complications including dental symptoms. Dental manifestation is characterized by early loss of primary anterior teeth. Diagnosis is based on a low concentration of ALP in serum. Enzymatic substitution therapy with recombinant ALP is available since 2015.
X-linked hypophosphatemia (XLH) is the most common form of rickets and osteomalacia. The disease manifests by growth retardation, deformities of lower limbs, bone and muscular pain. Dental manifestation is dentin dysplasia, followed by formation of periapical abscesses of intact teeth, mainly in primary dentition. A humanized monoclonal antibody for FGF23 (burosumab) is the promising treatment for XLH.
Úvod
Hypofosfatázie a X-vázaná hypofosfatemická křivice patří mezi vzácná dědičná metabolická onemocnění spojená s poruchou metabolismu vápníku a fosforu.
Hypofosfatázie (HPP) je onemocnění charakterizované dominujícím skeletálním postižením a mnohočetnými systémovými projevy v důsledku snížené koncentrace tkáňové nespecifické alkalické fosfatázy (ALP) v séru [1]. Vzniká v důsledku mutace v genu ALP lokalizovaného na prvním chromozomu. Dědičnost je nejčastěji autozomálně recesivní (těžší formy) nebo autozomálně dominantní (lehčí formy postižení) [2]. ALP se exprimuje v kostech, zubních tkáních, játrech, CNS, placentě a nadledvinách. Základní funkcí ALP je odštěpení fosfátů z různých molekul pomocí hydrolýzy. Význam ALP pro mineralizaci tkání tvořených hydroxyapatitem (kosti, tvrdé zubní tkáně, především zubní cement) je velmi důležitý. Konverzí anorganického pyrofosfátu, který inhibuje mineralizaci na fosfát, dochází k tvorbě hydroxyapatitu (HA), což je základní látka podmiňující pevnost kostí a tvrdých zubních tkání. Nezbytným předpokladem pro mineralizaci je přítomnost kolagenu v místě působení ALP, což vysvětluje, proč za fyziologických podmínek nedochází ke zvápenatění v játrech, mozku nebo placentě, ačkoli i zde se ALP exprimuje [3, 4].
X-vázaná hypofosfatemická křivice (XLH), dříve označovaná za vitamin D rezistentní křivici, je nejčastější typ vrozené, geneticky podmíněné křivice. Přenos je dominantní s vazbou na X chromozom a projevuje se hypofosfatemií, osteomalacií a růstovou retardací. Onemocnění je primárně způsobeno mutací v PHEX genu (phosphate regulating gene homologous to endopeptidases on the X chromosome), který je lokalizován na krátkém raménku chromozomu X a je primárně exprimován v buňkách kostí a tvrdých zubních tkání. Důležitou roli v patofyziologii XLH hraje fibroblastový růstový hormon 23 (FGF23), který je u těchto pacientů uvolňován z osteocytů. Zvýšená hladina FGF23 snižuje reabsorpci fosfátů v ledvinách. Renální ztráty fosforu vedou k jeho snížené hladině v séru, což poškozuje funkci osteoblastů a způsobuje poruchu osifikace [5, 6, 7, 16].
Epidemiologie
V obou případech jde o onemocnění s nízkou prevalencí. Závažné formy hypofosfatázie se u evropské populace vyskytují v počtu 1 případ na 300 000 živě narozených dětí, mírnější formy v poměru 1 : 6300. Častější výskyt hypofosfatázie byl zaznamenán v některých mimoevropských populacích, např. v Japonsku nebo v Kanadě [1, 8].
X-vázaná hypofosfatemická křivice se vyskytuje v počtu 1 případ na 20 000 – 60 000 živě narozených dětí. XLH se většinou dědí jako dominantní znak vázaný na chromozom X, ale u 20 – 30 % případů se vyskytuje spontánně s negativní rodinnou anamnézou [5, 9].
Klinická diagnostika
Hypofosfatázie je klinicky heterogenní onemocnění, vyskytující se v pěti základních formách [1]. Záměrně je vynechána forma adultní, která se u dětí nevyskytuje. Základní klinická charakteristika je uvedena v tabulce 1.
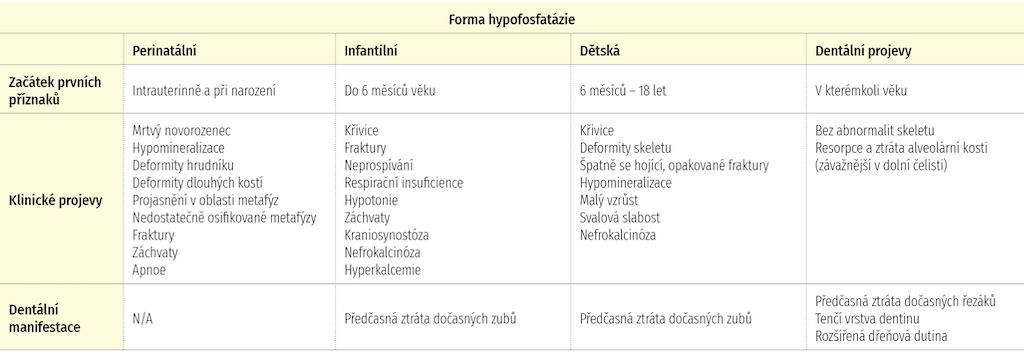{kind=link}
Ze skeletálních symptomů dominuje fragilita a deformace kostí, které se mohou vyskytovat již prenatálně nebo perinatálně. Hypofosfatázie diagnostikovaná v časném dětství vede k celkovému neprospívání dítěte a nedostatečnému růstu skeletu. U dětské formy HPP jsou častým nálezem obtíže s chůzí až neschopnost pohybu a pacienti jsou odkázáni na různé rehabilitační pomůcky nebo invalidní vozík. V objektivním nálezu jsou nápadné deformity hrudníku, široce otevřená velká fontanela a hypertelorismus. Současně se objevuje také svalová slabost. Dalšími příznaky bývají předčasné uzavírání lebečních švů za vzniku kraniosynostózy a tracheomalacie s tendencí k respiračním infektům. Častým laboratorním nálezem u batolat a předškolních dětí bývá hyperkalcemie. Komplikací kraniosynostózy bývá zvýšení intrakraniálního tlaku. Později se na skeletu objevují změny připomínající rachitidu – deformita hrudníku, rozšíření metafýzy dlouhých kostí a genua vara. Vývoj motoriky je celkově opožděn [10, 11, 12].
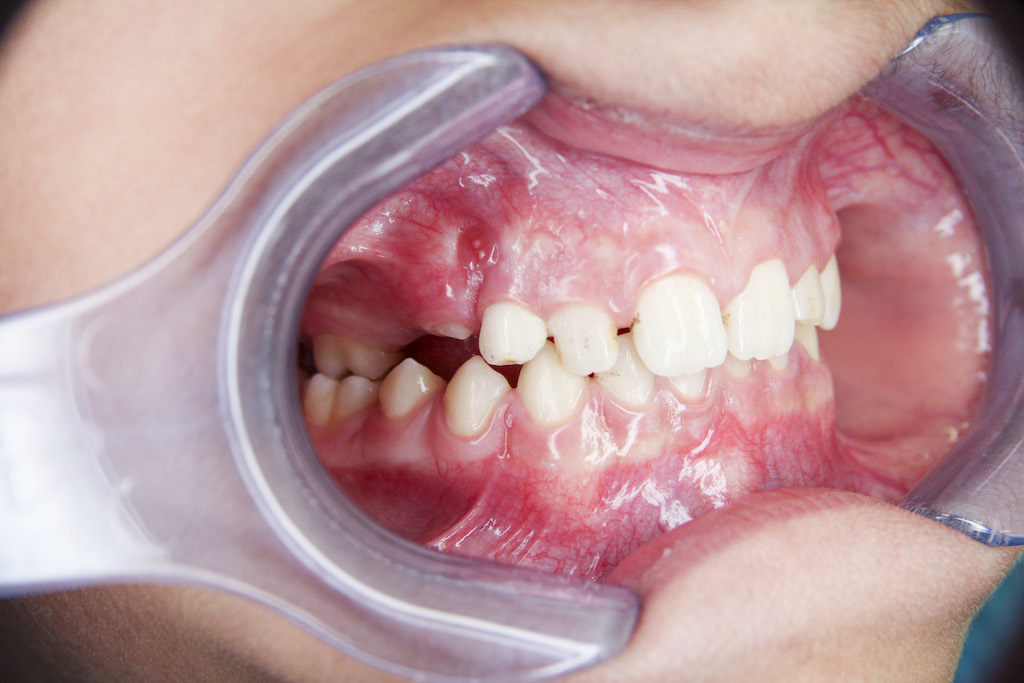
Z dentálních nálezů dominuje předčasná ztráta dočasných zubů mezi 3. a 4. rokem věku dítěte, typicky ve frontální krajině (obr. 1). Častěji a dříve bývají postiženy dočasné řezáky v dolní čelisti, později i v horní čelisti. Postižení ostatních zubů je značně variabilní [13].
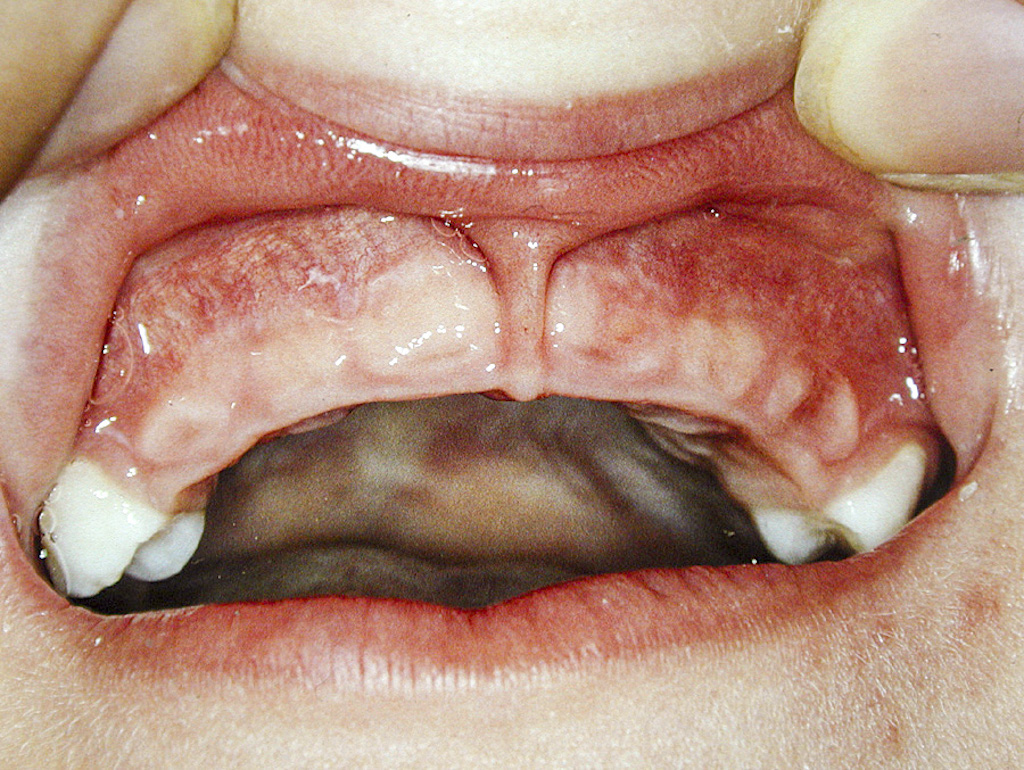{kind=link}
Předčasná ztráta zubů je důsledkem poruchy tvorby zubního cementu. Extracelulární akumulace anorganického pyrofosfátu může vést k poruše tvorby zubního cementu, který pokrývá kořeny zubů. Pokles hladiny alkalické fosfatázy vede k nedostatečné mineralizaci kostní tkáně a organického matrixu dentinu a cementu. Histopatologické vyšetření předčasně eliminovaných zubů prokázalo nedostatečnou tvorbu celulárního i acelulárního cementu. Dočasné zuby eliminují bez známky resorpce kořenů. Na rentgenovém nálezu je typická výrazná resorpce a ztráta alveolární kosti [13, 14, 18].
U mírných forem bývá předčasná ztráta dočasných zubů častým a také jediným projevem.
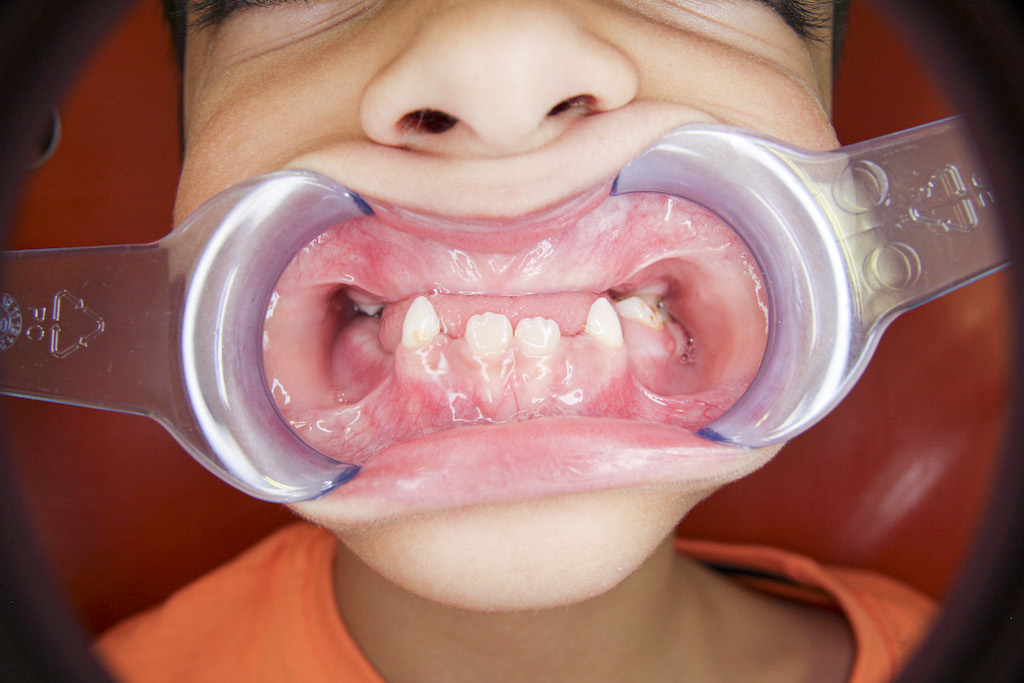
Předčasná ztráta zubů stálé dentice není typickým projevem HPP, ovšem u starších dětí a adolescentů k postižení parodontu může dojít. Mohou se objevit i poruchy tvorby dentinu a skloviny. Projevem dysplazie dentinu bývá rozšířená dřeňová dutina a velmi tenká vrstva dentinu (tzv. shell-teeth) (obr. 2). V případě skloviny to jsou nejčastěji kvantitativní poruchy ve smyslu hypoplazie, častěji u stálých zubů. Hyperkalcemie může vést k nechutenství a problémům s příjmem potravy, často řešeným nadměrnou konzumací volných cukrů v potravě, a tím ke zvýšenému riziku vzniku zubního kazu [10, 19].
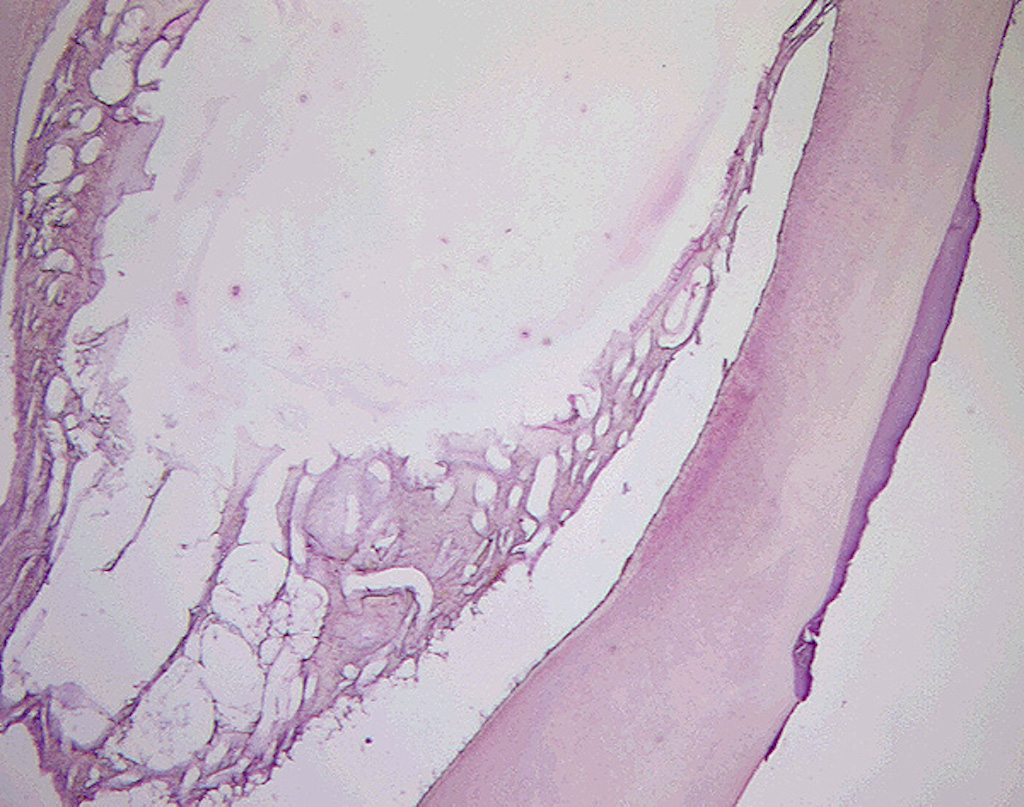{kind=link}
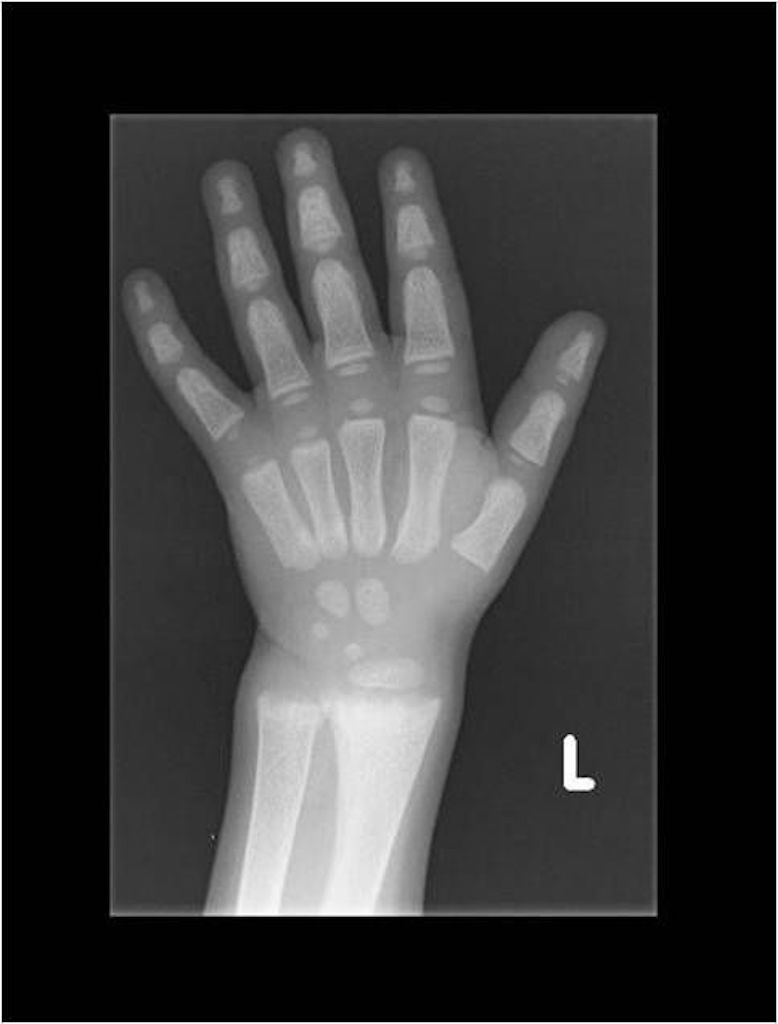
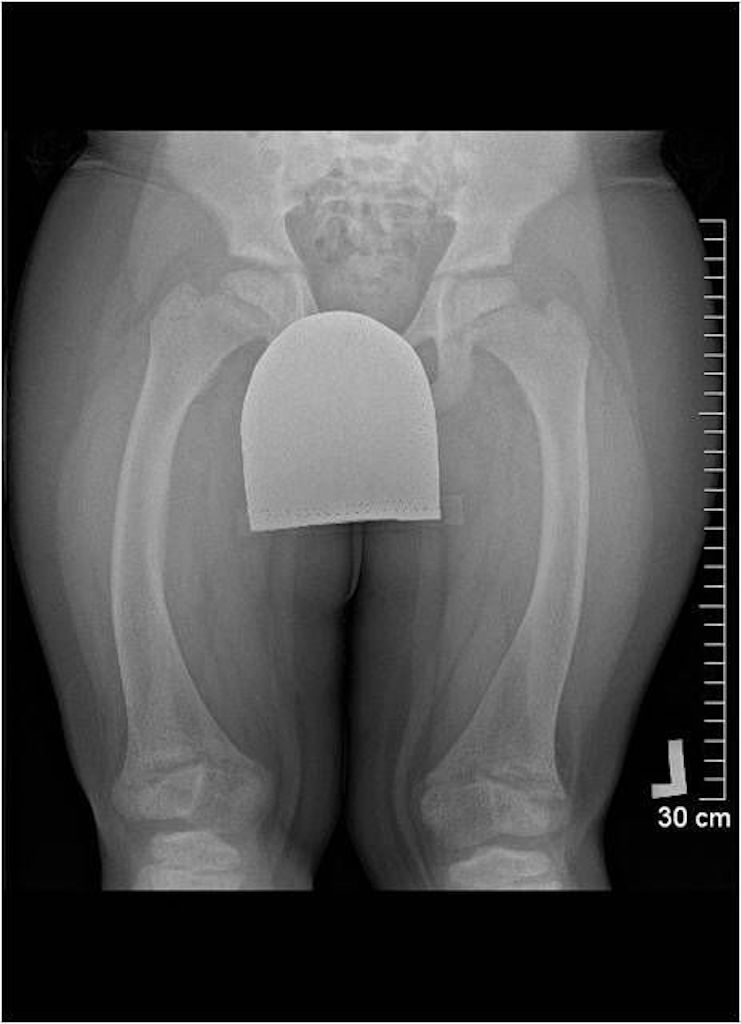
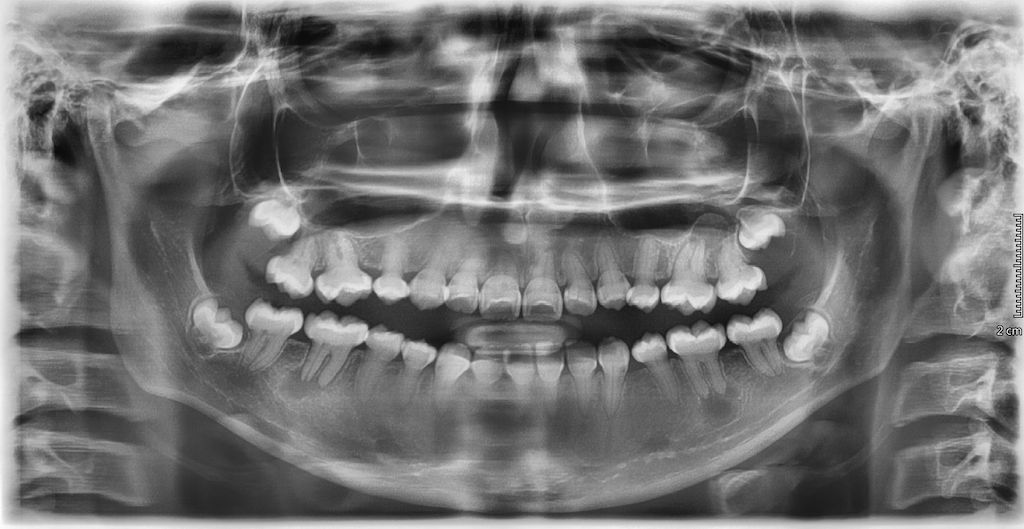
X-vázaná hypofosfatemická křivice (XLH) je vzácné onemocnění charakterizované značnou variabilitou v klinickém obraze (tab. 2), což může vést k pozdnímu rozpoznání choroby. Nejčastěji se onemocnění manifestuje v prvních dvou letech života zakřivením dolních končetin a opožděným růstem (obr. 3). Většina dětí má typické rachitické změny na rentgenovém snímku horních (obr. 4) a dolních končetin v oblasti kolen a částečně také v oblasti růstových plotének, což může vést k bolesti při chůzi. Nedostatečně osifikované kosti se začínají varózně ohýbat (obr. 5). Chůze postižených dětí je nejistá a kolébavá. Postižení axiálního skeletu je méně nápadné, kostní denzita páteře je většinou zachována. Pokud dítě není léčeno, jeho růst je disproporcionální, dolní končetiny jsou výrazně krátké vzhledem k trupu (obr. 6). K dalším fenotypovým charakteristikám patří deformity lebky ve smyslu dolichocefalie a kraniosynostózy [6, 14].
{kind=link}
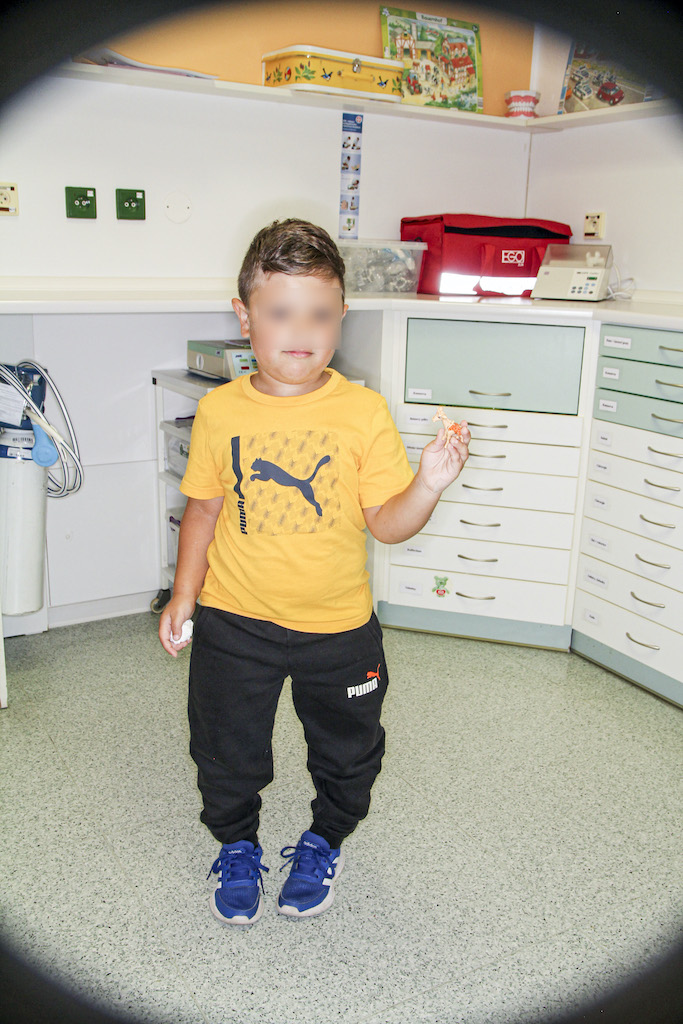{kind=link}
{kind=link}
{kind=link}
{kind=link}
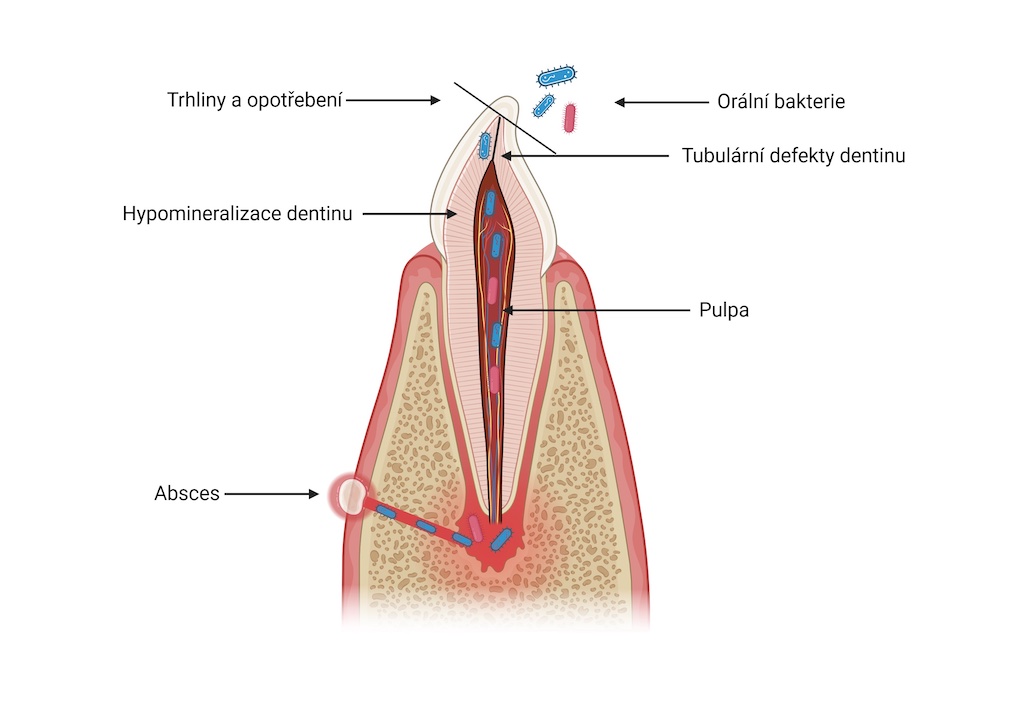
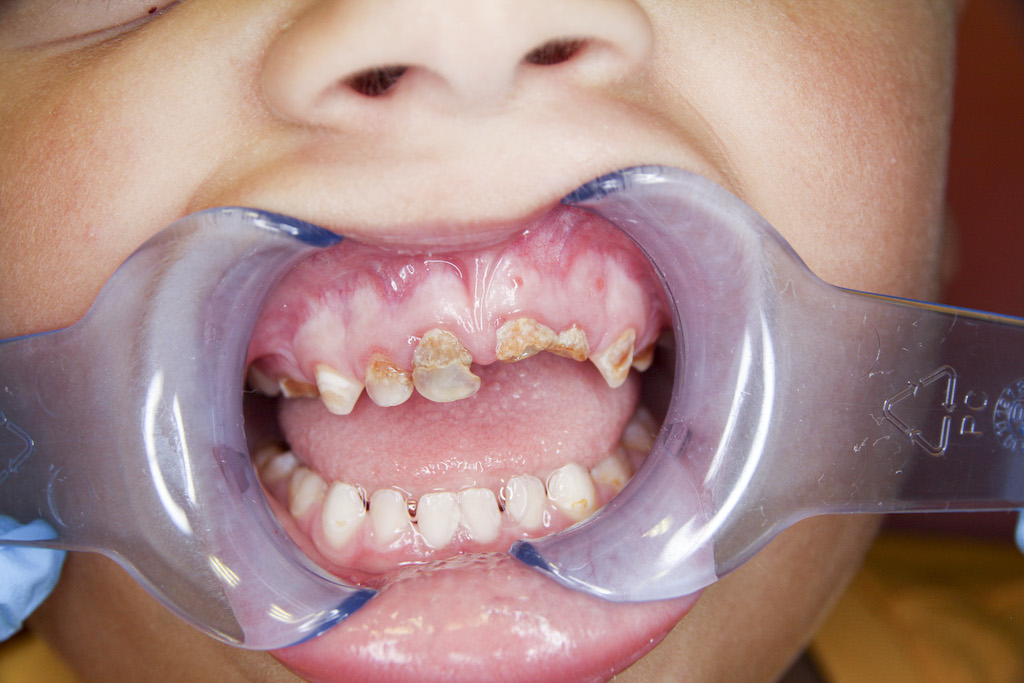
Z dentálních postižení dominuje porucha tvorby a mineralizace dentinu, což vede k tvorbě spontánních abscesů a píštělí u intaktních zubů a předčasné ztrátě zubů stálé dentice (obr. 7). Mikrofraktury nebo atrice skloviny umožňují infiltraci orálních bakterií přes hypomineralizovaný dentin do zubní dřeně. Následně dochází k nekróze zubní dřeně a periapikální patologii (obr. 8), (obr. 9). Dočasné zuby jsou postiženy častěji než zuby stálé dentice, pravděpodobně vzhledem k tenčí vrstvě skloviny a dentinu u dočasných zubů. Častěji bývají postiženy řezáky, zuby v laterálním úseku výjimečně (obr. 10). Na rentgenovém snímku je patrná velká dřeňová dutina, s výběžky směrem k incizní hraně a hrbolkům. Histopatologické vyšetření prokazuje širokou vrstvu predentinu a strukturální změny interglobulárního a tubulárního dentinu. Projevem dysplazie dentinu bývá opožděná erupce a krátké zaoblené zubní kořeny (obr. 11). Není známo, zda je riziko postižení zubním kazem u dětí s XLH vyšší v porovnání se zdravými dětmi [5, 17]. Progrese kariézního postižení je u dětí s XLH rychlejší vzhledem k tenké vrstvě skloviny a dysplazii dentinu. Mohou se objevit také poruchy okluze, nejčastěji otevřený skus [5, 13].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
V laboratorním obraze dominuje hypofosfatemie s hyperfosfaturií. Plasmatická koncentrace vápníku, ALP a vitaminu D bývají v normě. Při léčbě fosfátovými roztoky a vitaminem D jako konvenční terapii může dojít k rozvoji nefrokalcinózy a zhoršení renálních funkcí [17].
Terapie
Hypofosfatázie: v současné době je v ČR k dispozici rekombinantní ALP (Strensiq®), která výrazným způsobem zlepšuje prognózu onemocnění postižených dětí a tím kvalitu života dětských pacientů. Preparát se podává 3x týdně subkutánně. Dlouhodobé klinické zkušenosti s tímto preparátem jsou zatím omezené, ale studie z posledních let ukazují zlepšení stavu ve všech parametrech (muskuloskeletální, respirační, psychomotorická a neurologická symptomatologie) [1, 13, 18].
Standardní léčba X-vázané hypofosfatemické křivice (XLH) zahrnuje perorální podávání fosfátů a aktivního vitaminu D. Dávkování je individuální a závisí na věku, hmotnosti, závažnosti onemocnění a schopnosti dítěte tolerovat léčbu. Terapie by měla být zahájena co nejdříve po stanovení diagnózy. Tato léčba může být spojena s řadou komplikací. Mezi nejzávažnější patří hypervitaminóza D způsobující hyperkalcemii, hyperparathyreóza a nefrokalcinóza. Od roku 2019 je v ČR možná léčba protilátkou proti FGF23, burosumabem. Burosumab je rekombinantní lidská monoklonální protilátka (IgG1), která se váže na FGF23 a tlumí jeho aktivitu. Dochází ke zvýšení tubulární reabsorpce fosfátů v ledvinách, k normalizaci fosfatemie a ke zlepšení mineralizace kostí. Preparát se podává subkutánně každých 14 dní [6, 14].
Doporučení pro praktické zubní lékaře
Praktický zubní lékař by měl pravidelně vyšetřovat dětské pacienty od věku 12 měsíců a dále každých 6 měsíců. Může tak být lékařem, který první pojme podezření na tato metabolická onemocnění [6, 10, 18]. Zvýšené pozornosti a konzultace s praktickým lékařem pro děti a dorost je zapotřebí v situacích uvedených v tabulce 3.
{kind=link}
Závěr
Hypofosfatázie stejně jako X-vázaná hypofosfatemická křivice jsou závažná, ale v současné době léčitelná onemocnění. Včasné stanovení diagnózy a zahájení adekvátní terapie jsou zásadní pro zlepšení prognózy těchto pacientů a předcházení trvalému skeletálnímu postižení. Úloha lékařů, pečujících o dětské pacienty (pediatři, praktiční zubní lékaři) spočívá v pečlivém vyšetření a hodnocení klinických a laboratorních nálezů a v případě podezření na tato onemocnění ve včasném odeslání dítěte na specializované pracoviště, kde bude zahájena adekvátní terapie.
Podpořeno: Cooperatio Program, vědní oblast DENT a podpořeno MZ ČR – RVO (FNHK, 00179906).
Fotogalerie














Literatura
1. Šumník Z, Souček O, Lebl J. Hypofosfatázie: Kdy na ni myslet a jak ji léčit. Pediatr. praxi. 2016; 17(3): 146 – 149.
2. Mornet E. The tissue nonspecific alkaline phosphatase gene mutation database. www.sesep.uvsq.fr/03_hypo_mutations.php.
3. Okawa R, Nakano K, Matsumoto M. Oral manifestations of patients with hypophosphatasia. Pediatric Dental Journal. 2012; 22(2): 155 – 162.
4. Lezot F, Descroix V, Hotton D, et al. Vitamin D and tissue non-specific alkaline phosphatase in dental cells. Eur J Oral Sci. 2006; 114: 178 – 182.
5. Okawa R, Nakano K. Dental manifestations and oral management of X-linked hypophosphatemia. Endocrines. 2022; 3: 654 – 664.
6. Sabandal MM, Robotta P, Burklein S, Schafer E. Review of the dental implications of X-linked hypophosphaetemic rickets. Clin Oral Investig. 2015; 19: 759 – 768.
7. Nakanishi T, Michigami T. Pathogenesis of FGF23-related hypophosphatemic diseases including X-linked hypophosphatemia. Endocrines. 2022; 3: 303 – 316.
8. Mornet E. Hypophosphatasia. Orphanet J Rare Dis. 2007; 2(1): 40.
9. Skálová S, Rozsívalová P, Koberová Ivančaková R. X-vázaná hypofosfatémie u dvou sourozenců – možnosti a úskalí konvenční terapie. Pediatr.praxi. 2020; Suppl C: 21 – 25.
10. Bloch-Zupan A. Hypophosphatasia: diagnosis and clinical signs. Int J Paediatr Dent. 2016; 26: 426 – 438.
11. Rockman-Greenberg C. Hypophosphatasia. Pediatr Endocrinol Rev. 2013; 10: 380 – 388.
12. Whyte MP. Hypophosphatasia – aetiolopgy, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016; 12: 233 – 246.
13. Kiselnikova L, Vislobokova E, Voinova V. Dental manifestation of hypophosphatasia in children and the effect of enzyme replacement therapy on dental status. Clin Case Rep. 2020; 8: 911 – 918.
14. Štarha J. X-vázaná hypofosfatemie (XLH) v dětském věku. Pediatr. praxi. 2020; Suppl C: 5 – 7.
15. Duplan MB, Norcy EL, Courson F, Chaussian C. Dental and periodonta features and management in XLH children and adults. Int J Bone Frag. 2021; 1: 74 – 79.
16. Baroncelli GI, Angioloni M, Ninni E, et al. Prevalence and pathogenesis of dental and periodontal lesions in children with X-linked hypophosphatemic rickets. Eur J Paediatr Dent. 2006; 7: 61 – 66.
17. Baroncelli GI, Mora S. X-linked hypophosphatemic rickets: Multisystemic disorder in children requiring multidisciplinary management. Front Endocrinol. 2021; 12: 688309.
18. Feeney C, Stanford N, Lee S, Barry S. Hypophosphatasia and the importance of the general dental practitioner – a case series and discussion of upcoming treatments. Brit Dent J. 2018; 224(12): 937 – 943.
19. Rafelsen S, Johansson S, Raeder H, Bjerknes R. Hereditary hypophosphatemia in Norway: a retrospective population-based study of genotypes, phenotypes, and treatment complications. Eur J Endocrinol. 2015; 174(2): 125 – 136.
16. 12. 2023
LKS. 2023; 33(12): 224 – 229
Autoři:
Fotografie
- Archiv autorů
Rubrika:
Téma: